Machine learning methods and their biomedical applications
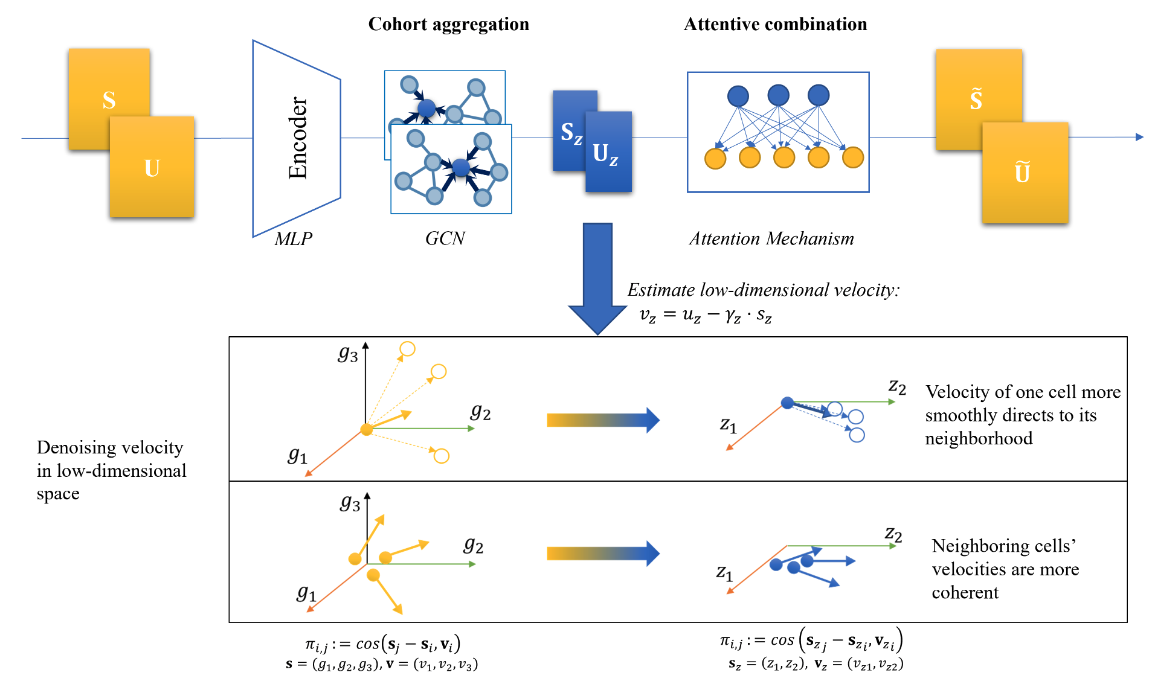
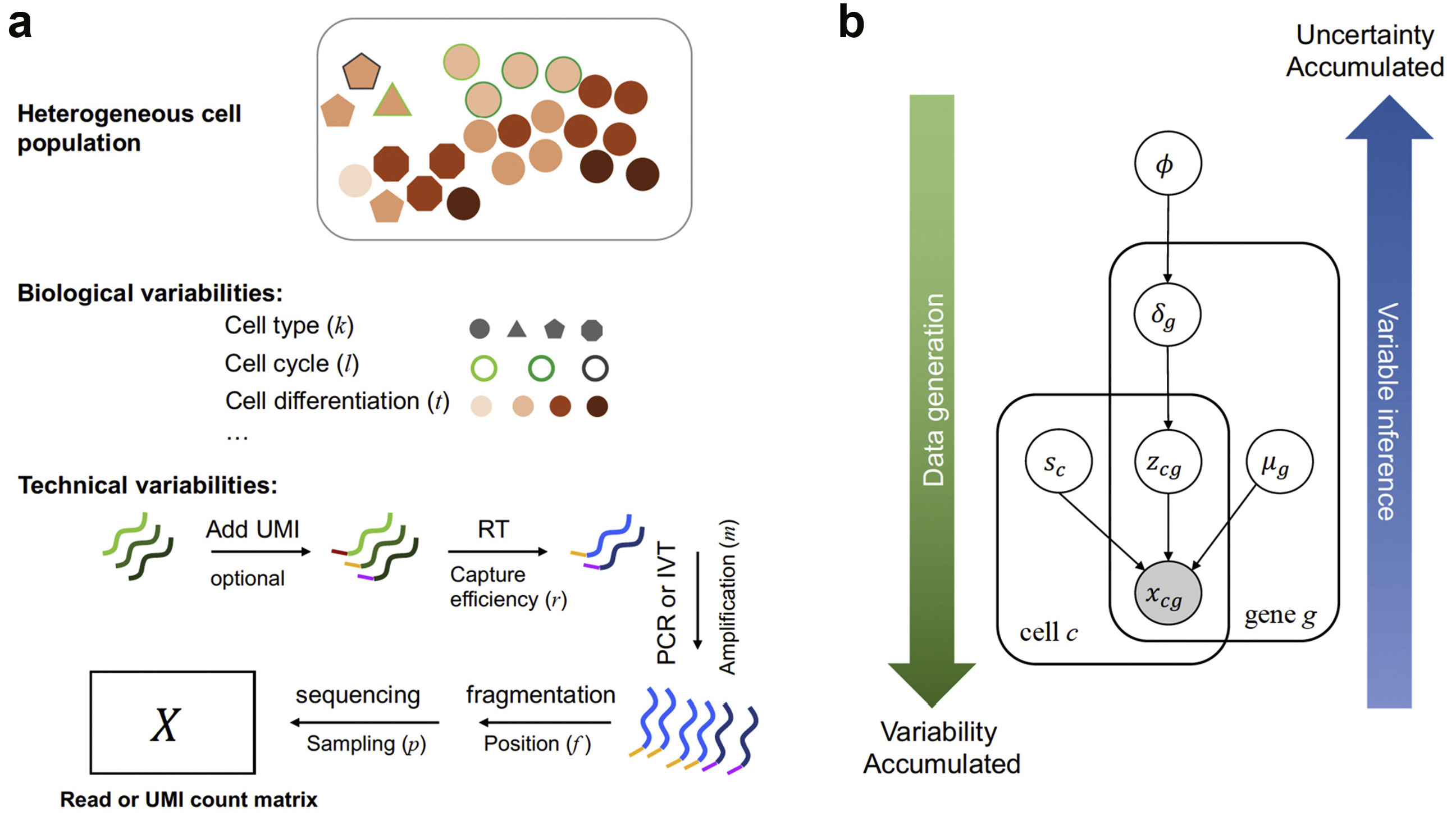
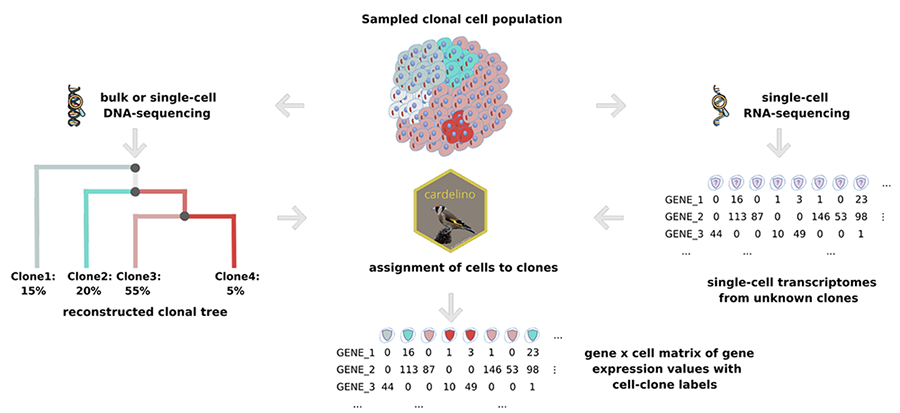
Our recent work covers both supervised learning and unsupervised learning, including predicting RNA splicing efficiency with genomic sequences and learning effective embeddings for robust RNA velocity analysis. We are further stretching into deep learning frameworks, often in conjunction with Bayesian graphical models.